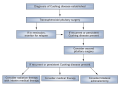
Management of Cushing DiseaseKey Points
- Transsphenoidal surgery is advisable for
most patients with newly diagnosed Cushing disease and often leads to
rapid and sustained remission of hypercortisolism with low morbidity in
most cases
- Pituitary reoperation might be
considered in patients with persistent or recurrent Cushing disease, but
the remission rate is lower in comparison to initial surgery
- Pituitary gland radiation therapy might
lead to remission in many patients with persistent or recurrent Cushing
disease, but takes a considerable time and a lifelong risk of
hypopituitarism exists
- Medical therapy can be used in patients
who are awaiting the therapeutic effects of pituitary gland radiation
therapy or in very ill patients in preparation for surgery
- Bilateral adrenalectomy could be
considered in patients with persistent or recurrent Cushing disease to
control hypercortisolism, but will induce adrenal insufficiency and
carries the risk of Nelson syndrome
Abstract
Cushing disease is caused by a corticotroph
tumor of the pituitary gland. Patients with Cushing disease are usually
treated with transsphenoidal surgery, as this approach leads to
remission in 70-90% of cases and is associated with low morbidity when
performed by experienced pituitary gland surgeons. Nonetheless, among
patients in postoperative remission, the risk of recurrence of Cushing
disease could reach 20-25% at 10 years after surgery. Patients with
persistent or recurrent Cushing disease might, therefore, benefit from a
second pituitary operation (which leads to remission in 50-70% of
cases), radiation therapy to the pituitary gland or bilateral
adrenalectomy. Remission after radiation therapy occurs in ~85% of
patients with Cushing disease after a considerable latency period.
Interim medical therapy is generally advisable after patients receive
radiation therapy because of the long latency period. Bilateral
adrenalectomy might be considered in patients who do not improve
following transsphenoidal surgery, particularly patients who are very
ill and require rapid control of hypercortisolism, or those wishing to
avoid the risk of hypopituitarism associated with radiation therapy.
Adrenalectomized patients require lifelong adrenal hormone replacement
and are at risk of Nelson syndrome. The development of medical therapies
with improved efficacy might influence the management of this
challenging condition.
Introduction Originally described by Harvey Cushing,
[1,2] Cushing disease is the most frequent cause of endogenous
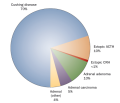
hypercortisolism (Figure 1). Cushing disease is caused by a tumor
originating from the corticotroph cells of the pituitary gland, or
rarely by corticotroph hyperplasia as a result of ectopic
corticotropin-releasing hormone (CRH) secretion, which leads to excess
adrenocorticotropic hormone (ACTH).
 (Enlarge Image)
(Enlarge Image)
Figure 1.
Causes of Endogenous Cushing Syndrome. Adrenal (other) includes
macronodular hyperplasia, McCune-Albright syndrome and primary pigmented
nodular adrenal disease (which might occur either in isolation or as a
manifestation of the Carney complex). Abbreviations: ACTH,
adrenocorticotropic hormone; CRH, corticotropin-releasing hormone.

Figure 1.
Causes of Endogenous Cushing Syndrome. Adrenal
(other) includes macronodular hyperplasia, McCune-Albright syndrome and
primary pigmented nodular adrenal disease (which might occur either in
isolation or as a manifestation of the Carney complex). Abbreviations:
ACTH, adrenocorticotropic hormone; CRH, corticotropin-releasing hormone.
Despite major advances in diagnosis and
therapy, Cushing disease is frequently a challenge for the clinician.
This Review summarizes data on the efficacy and safety of established
therapies for patients with Cushing disease and highlights agents that
are being investigated as possible future therapies for patients with
this condition. Although beyond the scope of the present article, it
should be noted that the management of the multiple comorbidities
associated with Cushing disease—including cardiovascular, metabolic,
catabolic, immunosuppressive and psychiatric complications—is very
important in optimizing patient outcomes.
[5]
Pituitary Surgery Transsphenoidal surgery is usually the
treatment of choice for most patients with Cushing disease, as it
typically leads to rapid and lasting remission of hypercortisolism while
preserving the function of the pituitary gland and adrenal glands in
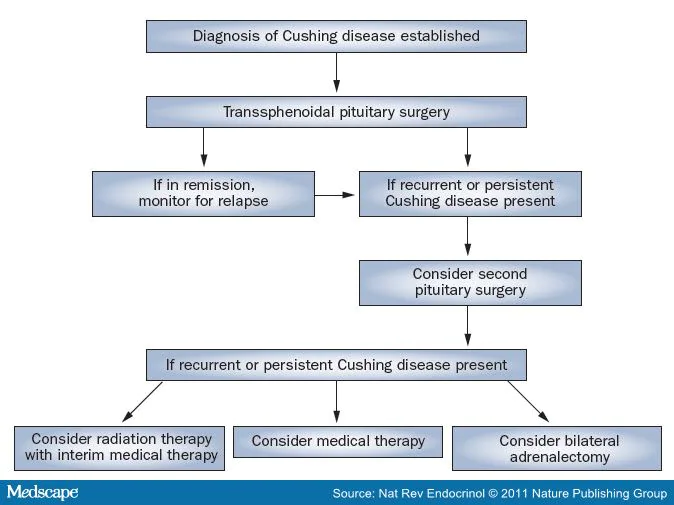
most patients (Figure 2).
[6] <blockquote>
</blockquote>
Figure 2.
Management of Patients with Cushing Disease.
Evaluation Before Surgery
Establishing the presence of pathological
hypercortisolism and elucidating its etiology are critical to patient
management. The diagnostic evaluation of patients with suspected Cushing
disease has been established (
Box 1).
[7] Patients with Cushing disease are at risk of infection and
cardiovascular events (both ischemic and thromboembolic). An evaluation
before surgery is warranted to identify patients at high risk of
perioperative complications and to optimize their overall health status
before the operation.
Surgical Technique In patients with Cushing disease, ~90% of pituitary gland tumors are microadenomas (<1 cm greatest diameter),
[8,9] and can be approached using the transsphenoidal route. This approach
can also be used to access most macroadenomas (≥1 cm greatest diameter).
Thus, the transsphenoidal approach is advisable in the majority of
patients with Cushing disease.
[10] Most surgeons approach the sphenoid
transnasally, usually by enlarging the ostium of the sphenoid sinus or
by making an incision at the junction between the sphenoid and the nasal
septum posteriorly. Extensive dissection to separate the nasal mucosa
from the septum is generally avoided, as this approach requires nasal
packing and is associated with a prolonged period for patient recovery.
The sublabial route to the sphenoid is no longer commonly used, because
this approach can lead to numbness and loss of sensation in the upper
lip and gums.
Fluoroscopy has been used for several decades
to guide the surgeon's approach to the pituitary gland. Frameless
stereotactic neuronavigation is the latest innovation and provides the
surgeon with helpful guidance when approaching the sphenoid and sella
turcica, which is particularly valuable in patients with variations in
their sphenoid anatomy or in patients undergoing repeat pituitary
surgery. Once the sphenoid sinus is entered, the sella turcica floor is
opened, exposing the dura mater. Exploration of the pituitary gland is
facilitated using either a fiber-optic endoscope or an operating
microscope. Biopsy samples are taken from suspicious areas within the
pituitary gland and submitted for frozen section examination by a
pathologist. If a tumor is identified during surgery, it is removed
using microsurgical instruments. If no tumor is visible, the sella
turcica is systematically explored, guided by preoperative imaging
findings as well as the lateralization data from bilateral inferior
petrosal sinus sampling (BIPSS).
[11,12] Some surgeons will empirically resect one half of the pituitary gland
on the side that shows lateralization on BIPSS, although this approach
has been shown to provide accurate information on tumor lateralization
before surgery in only around 65% of patients.
[11,12] Hemostasis is achieved using standard techniques (including packing or
bipolar cautery) and the resection cavity is frequently packed to
minimize the risk of cerebrospinal fluid leak after surgery. The floor
of the sella turcica is then reconstructed. Nasal packing is generally
not necessary with an endonasal approach.
Complications and Management After Surgery Transsphenoidal surgery of the pituitary
gland is associated with low perioperative mortality (≤1.5%),
approaching zero in several patient series.
[13] Surgical complications are less frequent in the hands of more
experienced surgeons compared to surgeons who perform this operation
less frequently, and might include cerebrospinal fluid leak (≤8%),
hemorrhage or hematomas (1-6%), epistaxis (0.4-6.0%), meningitis (≤3%)
and venous thromboembolism (≤4%). Endocrine complications might include
central diabetes insipidus (3-9%), hyponatremia as a result of
inappropriate secretion of antidiuretic hormone (10-25%) and
hypopituitarism (2-40%).
[13-15] Patients with Cushing disease who are
entering remission will probably experience hypoadrenalism for a number
of months after successful surgery as a result of suppression of the
hypothalamic-pituitary-adrenal (HPA) axis.
[16] During this period, glucocorticoid replacement is needed to prevent
symptomatic adrenal insufficiency. The timing of the initiation of
replacement therapy varies among different institutions. In some
hospitals, glucocorticoid replacement is not given during the early
period after surgery (typically 2-4 days). Instead, patients are
followed-up with clinical and biochemical assessments to detect evidence
of hypoadrenalism, which indicates biochemical remission of
hypercortisolism, before glucocorticoid replacement is begun.
[17,18] This practice requires prolonged inpatient monitoring, which might not
be logistically feasible in many hospitals. In other institutions
(including ours), therefore, patients are discharged early (24-36 h
after surgery) and are given a low (nonsuppressive) dose of
dexamethasone to prevent clinical hypoadrenalism while laboratory
testing takes place to determine whether biochemical remission has
occurred.
[15] Surgical Outcomes—remission Criteria Patients who are entering remission might
experience glucocorticoid withdrawal symptoms, even if glucocorticoid
replacement has been administered in the early period after surgery. In
some patients, supraphysiologic glucocorticoid doses might temporarily
need to be administered to prevent severe symptoms, including nausea or
vomiting, fatigue or diffuse achiness. For patients in remission,
withdrawal symptoms become gradually attenuated and the physical
manifestations of Cushing disease gradually wane over several months.
The laboratory tests and end points used to
determine whether remission of Cushing disease has occurred vary between
institutions and studies, and include very low early-morning serum
levels of cortisol or ACTH, low 24 h urine free cortisol levels, and
normal cortisol suppression in response to dexamethasone administration
(either 1 mg or 2 mg dexamethasone), measured in the first 2 weeks after
surgery.
[15,19-23] In a study published in 2008, low late-night salivary levels of
cortisol were proposed as a sensitive test to confirm remission.
[24] However, in contrast to the diagnosis of Cushing disease, for which
pertinent consensus criteria have been proposed, the criteria used to
establish remission of Cushing disease are not universally agreed upon,
which hinders direct comparison between outcome studies.
[7] Studies that cover a considerable amount of
surgical experience have been published that indicate that
transsphenoidal surgery might lead to remission of Cushing disease in
70-90% of patients (
Table 1). Factors suggested to influence the outcome of pituitary surgery
include the size and extent of the tumor, the identification of a tumor
in the sella turcica on an MRI scan before surgery, during the operation
or in pathology, and the surgeon's level of experience.
[13,15,20,21,25-28] The data from several studies indicate that
remission rates following surgery are higher among patients with
microadenomas than among those with macro-adenomas.
[15,23,29] In some, but not all, studies, higher remission rates have been
reported among patients for whom a tumor is visualized during an MRI
scan before surgery in comparison to those with no visible tumor on
imaging.
[25,26] Similarly, some studies have suggested that identification of a tumor by the surgeon during the operation
[27] or the presence of a pituitary gland adenoma on pathologic examination are associated with increased remission rates.
[20,28 However, there are clearly patients who have experienced lasting remission without an adenoma being confirmed by pathology.
[26,30] In these patients, it has been suggested that the small, fragile tissue
specimen might have not survived handling and processing. It is
presumed that the tumor was destroyed without obtaining a specimen for
analysis, so that the patients experience remission without a pathologic
diagnosis being established.
Some data indicate that the use of an
endoscopic approach to the sella turcica might lead to remission at a
rate comparable to that reported in studies that used a microscopic
approach.
[31] However, conversion from the endoscopic approach to the microscopic
approach might be needed in some patients (for example, if distorted
anatomy or mucosal bleeding obscure the surgeon's view).
[32] Additional data are required to compare outcomes of patients operated on using these two methods.
Patients thought to be in remission are
started on short-acting glucocorticoid replacement therapy (prednisone,
prednisolone or hydrocortisone) with regular, periodic reassessment of
early morning serum levels of cortisol measured 24 h after the most
recent dose of glucocorticoid to monitor for the recovery of the HPA
axis, which usually takes 6-12 months. Glucocorticoid replacement might
be safely discontinued when serum levels of cortisol (either at
baseline, measured in the morning, or after cosyntropin administration
to stimulate cortisol secretion) exceed 496.6 nmol/l. Lifelong follow-up
is needed in all patients with Cushing disease thought to be in
remission, as they are at long-term risk of relapse.
Evaluation for Disease Recurrence Several risk factors for relapse of Cushing
disease have been suggested that are based on results of testing in the
early period after the operation. An undetectable or low serum level of
cortisol in the early morning predicts a low risk of recurrence.
[18,33] However, a very low serum level of cortisol in the early morning does not universally predict long-lasting remission.
[30 Low plasma levels of ACTH also predict low risk of relapse.
[20] In addition, a prolonged (>1 year) requirement for glucocorticoid
replacement after pituitary surgery might indicate a low recurrence
risk.
[25,34] Factors suggested as being predictive of increased recurrence risk
include normal (rather than subnormal) serum levels of cortisol after
surgery,
[33,35] inadequate suppression of the HPA axis after administration of 1 mg dexamethasone
[36] or loperamide
[37] suppression testing, and excessive stimulation of the HPA axis on CRH,
[38] desmopressin,
[37] dexamethasone-suppressed desmopressin stimulation
[39] or metyrapone
[40] testing.
The available data indicate that the risk of recurrence of Cushing disease might reach 20-25% at 10 years after surgery (
Table 1).
[18,19] All patients initially thought to be in remission require lifelong
follow-up, including clinical and laboratory re-evaluation to examine
the possibility of recurrence of Cushing disease, which might occur even
20 years after surgery.
[41] Measuring 24 h urine free cortisol or performing a 1 mg dexamethasone
suppression test are used most frequently to detect disease recurrence.
In addition, measuring late-night salivary levels of cortisol is a
sensitive test in the diagnosis of recurrent Cushing disease.
[24,42] Re-evaluation of the diagnosis of Cushing
disease is advisable in all patients found to have disease recurrence,
as well as those with persistent hypercortisolism after pituitary
surgery. This re-evaluation is particularly important if further therapy
directed at the pituitary gland is being considered. Re-evaluation
should include review of laboratory data obtained before surgery to
ensure that the diagnosis of pathological, ACTH-dependent
hypercortisolism is accurate, as well as scrutinizing laboratory and
imaging studies that have pointed to the pituitary gland as the presumed
source of ACTH excess. If BIPSS was performed before surgery, imaging
and laboratory data obtained during this procedure should be reviewed
and discussed with the radiologist, if possible, to ensure that the
results are reliable and accurate. Of note, patients with the rare
syndrome of ectopic CRH excess, secreted from a tumor outside the
cranium, might show a central ACTH gradient on BIPSS (central to
peripheral plasma ACTH ratio exceeding 3:1).
[43] Review of the operative notes and pathology report from the pituitary
surgery are also advised, as the presence of an adenoma provides
diagnostic confirmation of Cushing disease. Even if an adenoma was not
identified after surgery, the clear presence of hypoadrenalism after
surgery provides presumptive evidence in support of a pituitary gland
source of ACTH excess.
[16]
Managing Persistence or Recurrence Patients with persistent or recurrent Cushing
disease need to be considered for additional therapies to minimize the
deleterious consequences of hypercortisolism. Possible treatments in
this group of patients might include a second transsphenoidal
exploration, radiation therapy, medical therapy or bilateral
adrenalectomy (Figure 2).
Pituitary Reoperation Pituitary reoperation might be considered in
patients with persistent or recurrent Cushing disease, particularly if
the first procedure was not performed by an experienced pituitary gland
surgeon. A second pituitary operation leads to remission in ~50-70% of
these patients.
[17,35] Early reoperation has been recommended in patients with persistent
Cushing disease after initial pituitary surgery, and might be indicated
in patients with persistently high or rising cortisol levels early in
the postoperative period.
[17] However, a delayed remission can occur in 5.6% of patients after the initial operation,
[44] suggesting that serial testing over a period of 1-2 months might be
advised in some patients who seem to be improving, but are not clearly
in remission immediately after surgery, before considering additional
therapy.
Overall remission rate after a second
pituitary operation is lower than after the first operation, and might
vary on the basis of patient characteristics, including the size and
location of the pituitary gland tumor.
[45] Pituitary reoperation is associated with a higher risk of anterior
hypopituitarism and cerebrospinal fluid leak compared to the first
operation.
[17] Patients with the rather uncommon ACTH-secreting macroadenomas might
not benefit from a second surgical procedure, particularly if tumor
invasion into the cavernous sinus or sphenoid was demonstrated at the
time of initial pituitary surgery.
Radiation Therapy With the exception of patients who are unfit
or decline surgery, radiation therapy is generally used as a second-line
option in patients with Cushing disease who do not respond to pituitary
surgery. Compared to surgery, the major caveats associated with
radiation therapy include the long delay between therapy and a
biochemical response and the considerable risk of hypopituitarism.
Techniques. The techniques used to
deliver radiation therapy to the pituitary gland continue to evolve.
Conventional fractionated radiation therapy has been used for decades to
treat patients with pituitary gland adenomas, including those with
Cushing disease.
[46,47] This approach involves administration of photons delivered through
several portals focused on the target tissue. The treatment is
administered in multiple small doses over a period of several (5-6)
weeks and generally leads to low-dose irradiation of brain tissue
outside the sella turcica.
In addition to conventional fractionated
photon beam radiation therapy, a number of approaches have been
developed to deliver precisely-focused (stereotactic) radiation therapy
to the pituitary gland, aimed at minimizing exposure to normal brain
tissue. Radiation beams can be delivered stereotactically using a
variety of equipment (Leksell Gamma Knife® [Elekta AB, Stockholm,
Sweden]
[48,49] or linear accelerator
[50,51] delivering photons and cyclotron delivering protons
[52,53]).
A stereotactic frame is secured to the patient's head to permit precise
positioning and imaging data are analyzed with computer algorithms to
plan delivery of radiation therapy in a narrow field. Proton beam
techniques, currently available in just a few centers worldwide, enable a
more selective delivery of radiation therapy in comparison to
approaches based on photon beams. In addition, proton beam techniques
considerably reduce radiation exposure to the normal brain tissue.
The term 'radiosurgery' has been used to
define radiation therapy delivered as a single, high dose in one
treatment session. However, stereotactic methods might also be used to
deliver radiation therapy in multiple fractions (several sessions of
low-dose radiation given over time), which is advisable for patients
with tumors close to the optic apparatus to decrease the risk of injury
to the visual pathway.
Outcomes and Complications.
Radiation
therapy is effective in controlling pathological hypercortisolism in up
to 86% of patients and prevents tumor growth in ~90-100% of patients (
Table 2).
[34,49,53,54] As is the case in patients with Cushing disease undergoing pituitary
surgery, the criteria used to establish remission of Cushing disease
after radiation therapy are not universally agreed upon, hindering
direct comparison between outcome studies.
A considerable delay (ranging from several
months to several years) exists in achieving a therapeutic effect with
radiation therapy, although this lag in response might be shortened if
radiosurgical methods are used. However, no trials exist that directly
compare these newer forms of radiation therapy to more conventional
approaches. Retrospective data indicated that a smaller treatment tissue
volume and absence of medical therapy at the time of Leksell Gamma
Knife® therapy are associated with improved patient outcomes.
[55] However, further studies are needed to examine whether medical therapy
might have a radioprotective effect, as has been suggested in patients
with acromegaly.
[56] Techniques for radiation therapy have
gradually become more refined, improving the accuracy of dose delivery,
which in turn has led to steeper dose gradients at the target perimeter,
thus minimizing exposure of normal tissues to radiation. Although it is
anticipated that these characteristics could translate into improved
overall safety, it is clear that adverse effects of radiation therapy
might still occur. In particular, the risk of hypopituitarism is ~30-40%
at 5 years after radiation therapy and probably increases over time.
[53,57] All patients who receive radiation therapy should be informed of the
risk of hypopituitarism and advised to have regular, lifelong evaluation
of the function of their pituitary gland. Uncommon adverse effects
include damage to optic nerves or chiasm,
[58] other cranial neuropathies,
[58] temporal-lobe necrosis,
[58] and secondary tumor formation.
[59,60] Although use of stereotactic techniques, careful target planning and
dose fractionation might decrease the risk of these adverse events,
patients still need to be informed of the possible risks involved.
Medical Therapy Medications being used or studied in patients
with Cushing disease include adrenal steroidogenesis inhibitors, which
block one or several steps in cortisol biosynthesis, as well as
centrally-acting agents, which inhibit ACTH secretion from pituitary
gland tumors (
Table 3). In addition, a glucocorticoid-receptor antagonist (mifepristone is
currently being investigated as a therapy for patients with Cushing
syndrome of various etiologies, including Cushing disease. Notably, none
of the agents currently available are approved by the FDA specifically
for the treatment of patients with Cushing disease.
Medical therapy is generally not considered as a first-line treatment for patients with Cushing disease.
[6] However, medical therapy is advisable for patients with Cushing disease
who have received radiation therapy but are awaiting its effects to
manifest. Patients who are very ill (for example, experiencing
infection, profound muscle weakness or psychosis) might also benefit
from medical therapy in preparation for surgery to ameliorate
hypercortisolism before surgery. In addition, medical therapy is a
therapeutic option for patients with ACTH-dependent hypercortisolism of
unclear origin or for those who are not candidates for surgery
(including patients with unstable cardiopulmonary status or uncontrolled
infection). Clearly, an unmet clinical need for highly efficacious and
safe medications for patients with Cushing disease exists. The
anticipated development of agents showing sustained efficacy and
acceptable safety profiles suggests that medical therapy might assume a
more prominent role in patient management in the future.
[61,62] Patients who receive medical therapy to control hypercortisolism
require meticulous clinical and laboratory monitoring for dose titration
and detection of hypoadrenalism. As an alternative to titrating medical
therapy to achieve normal cortisol levels, a 'block and replace'
regimen could be considered. This regimen involves use of larger
medication doses than the titration method to suppress endogenous
cortisol synthesis to below normal levels, while concomitant exogenous
physiologic glucocorticoid substitution is prescribed to prevent
hypoadrenalism.
[6] Physiologic glucocorticoid substitution might be more difficult to
implement in practice using the 'block and replace' regimen, but could
be useful in selected patients, including those with periodic
hormonogenesis (cyclic Cushing disease).
Steroidogenesis Inhibitors. Steroidogenesis
inhibitors are widely used to control hypercortisolism, but have no
effect on the size of the pituitary gland tumor. In some patients,
escape from their effects (secondary failure of the medication) might
occur as a result of increased secretion of ACTH by the pituitary gland
tumor in response to decreased feedback inhibition as a result of
lowered systemic cortisol levels.
Ketoconazole has been used extensively to decrease excess cortisol
levels by inhibiting several steroidogenic enzymes. Administered as a
monotherapy, ketoconazole decreases cortisol levels in ~70-80% of
patients.
[63] Monitoring of systemic levels of liver enzymes is advisable to detect
the development of abnormalities that might require dose adjustment or
drug discontinuation.
[64] Reversible abnormalities in liver enzymes occur in ~10% of patients,
but serious liver injury is much less frequent, occurring in ~1 of
15,000 patients.
[64,65] Metyrapone predominantly inhibits 11 β hydroxylase and has been used
either as a monotherapy, leading to a normalization of cortisol levels
in ~75-80% of patients, or in combination with other steroidogenesis
inhibitors, achieving even higher efficacy.
[66] In addition, combination therapy might improve tolerance to metyrapone,
preventing or attenuating its androgenic adverse effects in women and
mineralocorticoid-related adverse effects in patients of both sexes.
Metyrapone is the steroidogenesis inhibitor that is most frequently used
during pregnancy, though there are safety concerns about the use of
this drug in pregnant women.
[67,68] In the USA, metyrapone can only be obtained through the manufacturer on an individual patient-by-patient basis.
Mitotane inhibits multiple steroidogenic enzymes and has additional
adrenolytic activity at high doses, but displays a relatively slow onset
of action compared to other steroidogenesis inhibitors.
[69] Use of mitotane is further limited by dose-related gastrointestinal and neurologic adverse effects.
[69] In addition, mitotane is stored in adipose tissue for ~2 years after
administration ends, and can not be used in women contemplating
pregnancy within 5 years from discontinuation of the treatment.
[70] Etomidate has primarily been used to induce anesthesia, and is the
only parenteral steroidogenesis inhibitor available for treatment of
hypercortisolism.
[71,72] Etomidate provides rapid control of excess cortisol and has been used
in patients with severe hypercortisolism refractory to other therapies,
but requires meticulous monitoring to prevent excessive sedation.
[71,72] Aminoglutethimide is efficacious in ~45-50% of patients as a
monotherapy, but is no longer available in the USA for marketing
reasons.
[73] Trilostane is a weak steroidogenesis inhibitor with limited efficacy and poor tolerance, which has restricted its use.
[74] Centrally-acting Agents.
Cabergoline is a dopamine agonist that is approved by the FDA as a therapy for hyperprolactinemia.
[75] As dopamine receptors are expressed in a subset of pituitary gland
tumors in patients with Cushing disease, cabergoline has been evaluated
as a potential therapy in patients with this condition.
[76] In a study published in 2009, a response to treatment with cabergoline was present in 15 of 20 patients.
[77] However, only 40% of these patients had a sustained response to
cabergoline over a 2-year period. In another study, a complete response
to cabergoline therapy was present in 11 of 30 patients and a partial
response in four of the patients.
[78] However, only nine of the patients had a sustained response to cabergoline after a mean period of ~3 years.
The doses of cabergoline used in these studies were relatively high
(1-7 mg per week and 0.5-4.0 mg per week, respectively, compared to a
usual dose of 0.5-2.0 mg per week), raising concerns about long-term
safety.
[77,78] Two large studies have suggested that cabergoline used at high doses as
a treatment for patients with Parkinson disease is associated with
cardiac valvulopathy risk.
[79,80] Serial echocardiographic monitoring might be advisable in patients with
Cushing disease on long-term cabergoline therapy, particularly when
administered at high doses.
[81] Pasireotide (SOM 230, Novartis, Basel, Switzerland) is a somatostatin
receptor (SSR) ligand that engages multiple receptor isoforms,
including SSRs 1, 2, 3 and 5.
[82] As SSR5 is often expressed by pituitary gland tumors in patients with
Cushing disease, pasireotide is being investigated as a therapeutic
agent in patients with this condition.
[76] A phase II trial has been published in which pasireotide administration
over a 2-week period was associated with a decrease in hypercortisolism
in 22 of 29 patients, including 16% of patients who experienced a full
biochemical response (defined as normalization in 24 h urine free
cortisol level).
[83] In addition to gastrointestinal adverse effects, which are common in
patients treated with SSR ligands, adverse effects of pasireotide
included hyperglycemia, which was noted in approximately one-third of
patients.
[83] These
promising data on the efficacy of pasireotide have led to the
initiation of a phase III study, which is currently at the stage of data
analysis.
Combination therapy might be more effective than monotherapy in patients with persistent Cushing disease.
[84,85] In two studies published in 2010 that used cabergoline and ketoconazole
or the stepwise combination of pasireotide, cabergoline and
ketoconazole, a complete response (defined as normalization of 24 h
urine free cortisol level) was present in up to 66% and 88% of patients,
respectively, in the short-term (3-6 months).
[84,85] Several other medications have been evaluated as possible therapies
for patients with Cushing disease, and shown to have limited or no
efficacy, including bromocriptine (dopamine agonist); octreotide
(primarily SSR2 ligand); cyproheptadine (serotonin-receptor antagonist);
sodium valproate (antiepileptic; facilitates γ aminobutyric acid
pathways); and rosiglitazone or pioglitazone (peroxisome
proliferator-activated receptor γ agonists).
[61,86] Glucocorticoid Receptor Antagonist. Mifepristone
(RU 486, Roussel Uclaf SA, Romainville Cedex, France) is a type 2
glucocorticoid receptor and progesterone receptor antagonist that is
being developed by Corcept Therapeutics as a possible therapy for
pathological hypercortisolism of various etiologies, including Cushing
disease.
[62] Currently, there is limited published evidence suggesting that
mifepristone might be effective in patients with refractory Cushing
disease.
[87] Hypoadrenalism might occur in treated patients despite high serum
levels of cortisol, and close clinical monitoring will be needed if this
agent becomes available for this purpose.
[87] Adverse effects of mifepristone include hypokalemia, hypertension, endometrial hyperplasia and fetal loss.
[87] Bilateral Adrenalectomy Before refinements in pituitary surgery were
implemented, bilateral adrenalectomy was widely used as a primary
therapy in patients with Cushing disease. At present, bilateral
adrenalectomy has a secondary role and is reserved for patients who do
not respond to pituitary surgery.
[88,89] In this subpopulation of patients, the procedure is frequently
advocated for individuals refractory or intolerant of several
interventions, including radiation and medical therapy.
In addition, bilateral adrenalectomy has an
important role in very ill patients, who are likely to benefit from
rapid control of hypercortisolism, as well as patients who might wish to
avoid the risk of hypopituitarism that is associated with radiation
therapy, including those of reproductive age. The precise indications
for bilateral adrenalectomy vary among different institutions. In all
cases, a thorough discussion with the patient is advised on the benefits
and shortcomings of this procedure.
Technique. Bilateral adrenalectomy is
generally performed using a laparoscopic technique, which requires
several small incisions to establish a pneumoperitoneum using carbon
dioxide insufflation, and trocar ports for insertion of a camera and
surgical instruments.
[90] Partial mobilization of the liver and retraction of the right kidney
are required to expose the right adrenal gland, and mobilization of the
left colon, spleen and pancreas are needed to expose the left adrenal
gland. After the adrenal gland is exposed and mobilized, the adrenal
artery and vein are clipped and divided and the gland is placed in a
retrieval pouch. The procedure is then repeated on the contralateral
side.
Outcomes and Complications.
Bilateral adrenalectomy is effective in controlling hyper-cortisolism in almost all patients.
[91-93] Compared to an open approach, laparoscopic adrenalectomy is associated
with a shorter hospital stay, lower morbidity and reduced period of
rehabilitation.
[94] The risk of perioperative complications is ~9.5-12.0%, and might
include bleeding, organ injury, infection, need for conversion to open
adrenalectomy, persistent pain, hernia and deep vein thrombosis.
[95,96] The major shortcoming of bilateral
adrenalectomy is the development of permanent adrenal insufficiency,
leading to a lifelong requirement for glucocorticoid and
mineralo-corticoid replacement therapies to avert a life-threatening
adrenal crisis.
[93] In addition, patients with Cushing disease are at risk of Nelson syndrome after bilateral adrenalectomy.
[97] This complication occurs in 8-38% of adrenalectomized patients and
involves growth of the pituitary gland corticotroph tumor, presumably
occurring as a result of removal of negative feedback inhibition after
the adrenalectomy.
[97,98] Patients with no visible lesions in the sella turcica at the time of
bilateral adrenalectomy might be at reduced risk for Nelson syndrome.
[98] In patients with corticotroph tumor progression (Nelson syndrome),
pituitary gland tumors might become locally invasive. In addition, a
substantial rise in systemic levels of ACTH might occur, causing skin
and mucosal hyperpigmentation. These pituitary gland tumors might
occasionally be amenable to resection, and usually respond well to
radiation therapy.
[53,99,100] Another potential long-term complication of bilateral adrenalectomy
involves the enlargement of either eutopic or ectopic adrenal rests
under ACTH stimulation, leading to recurrence of hypercortisolism.
[93]
Conclusions Transsphenoidal surgery of the pituitary
gland is usually the treatment of choice for patients with Cushing
disease. Patients with persistent or recurrent disease after pituitary
surgery could be offered a second pituitary operation, radiation therapy
to the pituitary gland with interim medical therapy, or bilateral
adrenalectomy.
Despite advances in techniques used in
surgery and radiation therapy, there are considerable unmet medical
needs in the treatment of patients with Cushing disease. The development
of new therapeutic agents is eagerly anticipated and might eventually
lead to paradigm shifts in the management of patients with this
challenging condition.